Frequently Asked Questions
What is LENS?
LENS (Landscape of Effective Neoantigens Software) is a workflow designed for predicting potentially therapuetically targetable tumor-specific and tumor-associated antigens from short-read sequencing data.
What antigen sources are supported?
Somatic single nucleotide variants (SNVs)
Somatic insertion and deletion variants (InDels)
Splice variants
Fusion events
Viruses
Endgoenous Retroviruses (ERVs)
Cancer-testis antigens (CTAs)
What is RAFT? How is it different from LENS?
RAFT is a nextflow-based workflow manager that runs LENS. RAFT is used to run
the LENS workflow, but RAFT is also capable of running other workflows (see
raft.py available-workflows). More information about RAFT can be found in
the RAFT Documentation.
What input files are required?
Users must provide a manifest file (see Manifest) as well as multiple samples per patient. These samples include:
DNA normal sample (WES/WXS or WGS)
DNA tumor sample (WES/WXS or WGS)
RNA tumor sample
Optionally, a patient-matched, tissue-matched RNA normal sample can be provided. If such a sample is not available, then a non-patient-matched, tissue-specific (e.g. GTEx) sample can be provided.
What sequencing technologies are supported?
Illumina-based short read (50 bp - 150 bp) sequencing is currently supported. A long-read based tumor antigen workflow is in development.
How can I run LENS with my own reference files?
Yes! See the Usage page for more information.
How can I run LENS using different tools?
Yes! RAFT allows users to utilize alternative (supported) tools by changing the
tool parameters within main.nf. See the Usage page for more
information.
How do I know which tool versions are running?
Tool versions are available in the Technical Details page.
How do I know which Docker iamge tools are running?
RAFT (the workflow manager running LENS) relies upon containerization software
(Docker or Singularity/Apptainer) for running its processes. The specific
container being used for each process can be found in your LENS project’s
nextflow.config file
(/path/to/raft/projects/<PROJECT_ID>/workflow/nextflow.config). Each
process defined within RAFT’s modules (the directories in workflow/) has
one or more Nextflow label directives assigned to it. For example the
fastp process includes the label fastp_container. If we search for this
label within the nextflow.config file then we find the lines:
This reveals that the fastp process is being run using BioContainer’s
fastp v0.23.1 Docker image.
How do I change which tool versions are used?
Tool verions can be changed by utilizing an alternative Docker image for
processes being run by a specific tool. Docker images can be found either on
DockerHub or BioContainers.
The tool’s corresponding process labels will need to be modified in the nextflow.config file
(/path/to/raft/projects/<PROJECT_ID>/workflow/nextflow.config).
Note
Docker images may differ in construction (e.g. where binaries are located
within the container), so some modifications to process definitions within
each tool’s RAFT module (in /path/to/raft/projects/<PROJECT_ID>/workflow/) may
be required to the corresponding process definitions.
How do I change resource allocations for each tool?
Resource allocations (cpus and memory directives) can be modified in
the nextflow.config file
(/path/to/raft/projects/<PROJECT_ID>/workflow/nextflow.config).
How is the workflow code in LENS executed?
See Technical Details for more information.
How is CTA gene list defined?
The CTA (cancer-testis antigens) gene list
(cta_and_self_antigen.homo_sapiens.gene_list) consists of CTAs defined
referencing the CTDatabase (http://www.cta.lncc.br/) list of genes. Note that
some genes have been added to the CTDatabase base set.
Why are some of my CTA transcripts expressed in normal tissues?
The CTAs defined in the CTDatabase reference list are not necessarily
testis-specific. To address this, LENS outputs contains columns that can be
used to further filter CTA pMHCs (such as gene_detectable_normal_tissues
defined using Human Protein Atlas data).
What do the mTEC columns mean?
Medullary thymic epithelial cells (mTECs) are cells within the thymus involved in central tolerance (https://en.wikipedia.org/wiki/Medullary_thymic_epithelial_cells). LENS utilizes two mTEC expression datasets (Larouche et al, Genome Medicine, 2020 and Laumont et al, Science Translational Medicine, 2018) to estimate CTA expression within mTEC tissues. Briefly, lower mTEC expression suggests a reduced risk of central tolerance towards the pMHC of interest.
How is ERV list defined?
The endogenous retrovirus (ERV) list is defined by the gEVE database version
1.1 (http://geve.med.u-tokai.ac.jp/). Note that the endogenous retroviral
elements contained within the gEVE database are computatoinally predicted. The
LENS report includes columns that can be used to further filter ERV pMHCs based
on metadata contained within the erv_scores.25SEP2023.tsv metadata file.
More information on these ERV scores can be found in the ERV section of the
Technical Details page.
How is CCF calculated?
Cancer cell fraction (CCF, also known as clonality) is estimated using the formula:
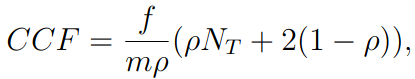
where f is the variant allele frequency, $rho$ is the sample tumor purity,
N_T is the gene-level copy number, and m is the multiplicity (Tarabichi
et al., 2021).
How is the priorization score calculated?
Tumor antigens are prioritized based upon the binding affinity, allele-specific transcript abundance, and CFF (if available), which aligns with previously published recommendations cite{wells2020key}.
Specifically, binding affinity values are transformed using:
$$ frac{abs(chi - 1000)}{1000} $$
such that higher affinity interactions with smaller nanomolar values will have larger transformed values while maintaining a distribution mirroring the original.
The support read counts will be log2 transformed and normalized by dividing each observation by the maximum observed count such that they range from $[0, 1]$.
The cancer cell fraction distributions will not require this normalization as they are already bound between $[0, 1]$. Next, each peptide and HLA allele combination will be assigned a prioritization metric using:
$$ S = pMHC_{BA} * pMHC_{RS}* pMHC_{CCF} $$
where $pMHC_{BA}$ is the transformed binding affinity, $pMHC_{RS}$ is the log-transformed and normalized read support, and $pMHC_{CCF}$ is the estimated cancer cell fraction.
This metric can be used to prioritize potential targets for manufacture and application to the patient’s tumor.
How are SNV and InDel peptides generated?
Answer coming soon!
How should I interpret the LENS report?
Answer coming soon!
How do I determine how many pMHCs are filtered at each step of LENS?
Answer coming soon!